
GUILHERME COSTA, VINÍCIUS CABRERA, VITOR ALMEIDA
 Foto: Cápsulas de fosfoetanolamina sintética – Bigstock
Foto: Cápsulas de fosfoetanolamina sintética – Bigstock
No fim de novembro, duas das maiores empresas do ramo farmacêutico, os laboratórios Pfizer, fabricante do Viagra, e Allergan, fabricante do Botox e remédios para o mal de Alzheimer fecharam um acordo de fusão estimado em cerca de US$160 bilhões, criando a maior empresa farmacêutica do mundo. Segundo a Folha de S. Paulo, a estimativa de vendas da nova companhia, Pfizer PLC, é de US$60 bilhões por ano. É a terceira maior operação de fusão e aquisição da história, atrás apenas das fusões Time Warner/America Online e Manesmann/Vodafone AirTouch.
Enquanto isso no Brasil a polêmica sobre a fosfoetanolamina sintética, a “pílula do câncer” que é produzida por menos de R$ 0,10 a unidade, está fora dos holofotes das grandes empresas farmacêuticas, mas sob os olhares atentos da população, instituições e governo. Em busca de respostas onde a Ciência ainda não chegou por completo, muitos pacientes com câncer e seus familiares buscam acesso à substância, à qual é creditada a potencialidade de tratar e até curar vários tipos de câncer, segundo relatos de usuários que possuem a doença. Entretanto, as etapas científicas que vão desde testes em laboratório até estudos clínicos ainda não chegaram à uma conclusão sobre a eficácia do potencial medicamento, o que reacende o debate sobre as consequências do uso de medicamentos não testados.
Histórico
A produção da substância teve início nas mãos do químico Gilberto Orivaldo Chierice, professor aposentado do Institudo de Química da USP em São Carlos (IQSC), no interior de São Paulo, que começou a estudar a hipótese da ação anticancerígena substância na década de 1980. Gilberto desenvolveu um método próprio para sintetizar a substância em laboratório. Porém, um processo oficial que comprove as propriedades terapêuticas do componente nunca foi realizado.
Em entrevista ao portal G1, Chiarice afirmou que na época as cápsulas começaram a ser administradas a pacientes com câncer do Hospital Amaral Carvalho de Jaú, também no interior de São Paulo, devido a um convênio do hospital com o IQSC. Porém, segundo o hospital, não há documentação que comprove a utilização dessas cápsulas por pacientes. Segundo ele, a divulgação da “cápsula da USP” começou quando “algumas pessoas tiveram melhora” e, mesmo depois de o hospital deixar de oferecer a substância aos pacientes, alguns familiares continuaram indo até o IQSC para pedir novas doses. O instituto, então, se manteve produzindo para atender a essa demanda, e em 2013 chegou a produzir mais de 50 mil cápsulas por mês. Depois da aposentadoria de Chiarice do IQSC, em 2013, o instituto interrompeu a produção da cápsula. Atualmente, por força de demandas judiciais de pacientes que conseguiram na Justiça o direito de receberem a substância, o instituto voltou a produzir e fornecer o produto.
“O paciente melhorava e recomendava para o vizinho. No início, a gente nunca controlou isso, não éramos médicos. Eles diziam: ‘Queria levar para a minha prima porque deve fazer bem’. Nossa atitude não era policiar isso”, afirma Chiarice. A dose tomada pelos pacientes não era controlada, e as orientações para o uso eram geralmente baseadas nas observações de parentes ou vizinhos.
Porém, é importante lembrar que “a palavra ´câncer ´ diz respeito a várias doenças diferentes, com sintomas e prognóstico diferentes e tratamentos diferentes” afirma Diogo Bugano, oncologista clínico do Hospital Israelita Albert Einstein. “Mesmo dois pacientes com o mesmo tipo de câncer podem não receber o mesmo tratamento, de acordo com o estadiamento da doença e com as outras condições de saúde que o paciente tem. Assim, o tratamento de um câncer de mama localizado apenas na mama é diferente do tratamento do câncer de mama que se espalhou para linfonodos ou para o fígado, que é diferente do tratamento do câncer de intestino que se espalhou para linfonodos ou para o fígado”, explica o oncologista.
O Conselho Regional de Farmácia de São Paulo (CRF-SP) chegou a autuar a Universidade de São Paulo (USP) por conta da produção da fosfoetanolamina sintética. Em vistoria realizada em outubro de 2015, fiscais estiveram no laboratório e verificaram que o processo não segue “boas práticas de produção” e não há controle de qualidade, incluindo um farmacêutico responsável pela fabricação e dispensação, o que levou à aplicação de uma multa e a um comunicado à Vigilância Sanitária (Anvisa). Entretanto, o Conselho não tem poder de interditar o laboratório, por ser uma fiscalização de caráter administrativo, afirmou Pedro Eduardo Menegasso, presidente e conselheiro do CRF-SP, em entrevista ao G1.
Segundo nota de esclarecimento oficial da Anvisa publicada no site, a Agência não recebeu qualquer pedido de avaliação para registro de medicamento referente à fosfoetanolamina, e nem pedido de pesquisa clínica, que é a avaliação com humanos. Isto significa que não há nenhuma avaliação de segurança e eficácia do produto realizada com o rigor necessário para a sua validação como medicamento com base nos critérios científicos aceitos mundialmente. Já uma segunda nota técnica diz que “A comercialização, bem como a exposição do produto fosfoetanolamina, estaria em desacordo ao que prevê a Lei nº. 6.360/76, que em seu artigo 12 assim dispõe: ´[…] nenhum dos produtos de que trata esta Lei, inclusive os importados, poderá ser industrializado, exposto à venda ou entregue ao consumo antes de registrado […]´”, diz o documento.
A distribuição da substância na USP chegou a ser cancelada devido à suspensão das liminares que autorizavam o envio da fosfoetanolamina sintética para pacientes com câncer. Entretanto, o presidente do Tribunal de Justiça de São Paulo, desembargador José Renato Nalini, reconsiderou o pedido baseado no entendimento do Supremo Tribunal Federal (STF) e abriu a prerrogativa para obtenção das cápsulas através da entrada de um pedido junto à justiça. O juiz destacou em sua decisão que não se podem ignorar os relatos de pacientes que apontam melhora no quadro clínico.
“Pondo-se de parte a questão médica, que se refere à avaliação da melhora, do ponto de vista jurídico há uma real contraposição de princípios fundamentais. De um lado, está a necessidade de resguardo da legalidade e da segurança dos procedimentos que tornam possível a comercialização no Brasil de medicamentos seguros. Por outro, há necessidade de proteção do direito à saúde”, afirmou o juiz. Portanto, qualquer brasileiro acometido pelo câncer pode acionar a justiça representada por advogado, o qual fará o pedido judicial. Para ingressar com a ação, são necessários documentos de identidade (RG, CPF e comprovante de residência) além de laudos médicos e exames que comprovem o diagnóstico da doença.
Os receios dos profissionais de oncologia, porém, não se restringem apenas à falta de conhecimento sobre os efeitos da substância. “Creio que existem 4 receios quanto ao uso de tratamentos alternativos não testados. O primeiro é o risco de serem danosos ao paciente. Assim como qualquer remédio remédio, um suplemento, chá ou outros compostos que o paciente toma podem ter efeitos colaterais. Existem vários relatos de hepatites graves causadas por chás e outros chamados ´remédios naturais´. O segundo é que estes tratamentos podem interagir com outros tratamentos tradicionais que o paciente recebe, diminuindo o efeito ou causando mais toxicidade. O terceiro é que pacientes podem abandonar tratamentos habituais, com eficácia comprovada, e preferir tratamentos alternativos. O quarto é a questão financeira: muitos destes tratamentos alternativos são caros. Quem já acompanhou um familiar com câncer sabe que o tratamento envolve muitos custos. Mesmo que a quimioterapia e consultas sejam pagas pelo SUS ou por um convênio, existem muitos gastos com exames, remédios, viagens para tratamento ou consulta e o custo dos dias de trabalho perdidos. Acrescentar a tudo isso o custo de um tratamento alternativo, que pode ou não funcionar e que pode ou não ser danoso ao paciente, pode gerar mais um sofrimento à estas famílias”, reitera o oncologista Diogo Bugano.
A Substância
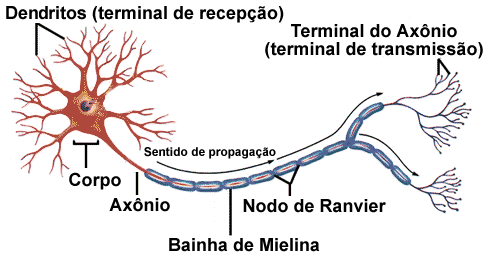
A Fosfoetanolamina é um derivado da etanolamina usada na formação de um tipo específico de esfingolipídio, a esfingomielina. A esfingomielina representa em torno de 85% de todos esfingolipídios no corpo humano e está presente naturalmente nas membranas plasmáticas celulares animais. É abundante no tecido nervoso, onde é especialmente encontrada na bainha de mielina que envolve o axônio de células nervosas, mas também pode ser identificada em glóbulos vermelhos e no cristalino dos olhos.

Ilustração: Fosfoetanolamina compondo uma molécula de esfingomielina – LHcheM
Segundo Salvador Claro Neto, pesquisador da USP, um dos detentores da patente da substância, nos anos 1990 integrantes do IQSC decidiram rever a literatura sobre a fosfoetanolamina. Apesar de os primeiros estudos sobre a substância datarem dos anos 1930 e indicarem grande presença da substância em tumores bovinos, o que apontava que talvez ela pudesse causar câncer, pesquisas mais recentes relacionavam a fosfoetanolamina à defesa do corpo. O organismo aumentaria a produção para combater o tumor – “e os cientistas do IQSC verificaram esta propriedade” – disse em entrevista ao G1.
“Toda célula cancerosa tem três características: citoplasma ácido, DNA modificado e pouco ATP, ou seja, pouca energia. Na célula, os produtores de energia são a mitocôndria e o citoplasma. Eles são como fábricas, quando uma falha, liga a outra. Se a mitocôndria começa a falhar, dispara o citoplasma, mas aí ele fica ácido, então se a mitocôndria não está produzindo energia aumenta a acidez. Isso muda as características químicas da célula e isso altera o DNA”, explica Neto. “A ideia é: a mitocôndria não produz energia porque não chega ácido graxo, e a fosfoetanolamina leva o ácido graxo”.
“Quando a pessoa ingere a fosfoetanolamina sintética, ela age como a natural e estimula o transporte de gordura. Com isso, as mitocôndrias que não estavam produzindo voltam a produzir e as células que já estavam defeituosas, com as características alteradas, sinalizam sua morte para o sistema de defesa, que as remove do organismo. É como se a célula estivesse trabalhando no 220 V. Quando a mitocôndria para de produzir, cai para 110 V e, quando vem a fosfoetanolamina, volta para 220 V. Aí dá curto, queima, e a célula que estava ácida e com DNA modificado sinaliza sua morte”, diz o pesquisador.
De acordo com artigo publicado na revista International Journal of Cancer Research and Treatment, a ação da fosfoetanolamina sintética (também conhecida como Pho-s) sugere que ela tem potencial de combate ao câncer. Em resumo, a molécula induziu citotoxicidade (nocividade à célula) em todas as células cancerosas estudadas sem afetar as células normais. Além disso, induziu a apoptose (morte celular programada) em células de tumores, inibiu o crescimento de tumores e aumentou a o tempo de vida de ratos. Uma pesquisa mais recente, publicada no British Journal of Cancer também aponta que a Pho-s combate células de leucemia tanto in vitro (células isoladas), quanto in vivo (em organismos vivos).
“Gostaríamos que a fosfoetanolamina sintética fosse produzida, disponibilizada pelo SUS. Procuramos os órgãos responsáveis, mas encontramos dificuldades porque não há protocolo clínico”, completa Neto.
 Infográfico: Ação da fosfoetanolamina sintética no organismo – Jornal de Santa Catarina
Infográfico: Ação da fosfoetanolamina sintética no organismo – Jornal de Santa Catarina
Aprendendo com o passado
Algumas drogas que se mostram promissoras em animais durante estudos em laboratório podem falhar ou causar consequências irreversíveis quando usadas em seres humanos. A Talidomida, por exemplo, foi um caso que levantou um intenso debate mundial sobre a segurança dos medicamentos. A substância apareceu no mercado pela primeira vez na Alemanha em 1957, e foi comercializada como um sedativo e hipnótico com poucos efeitos colaterais, destinado a combater a ansiedade, insônia e tensão. Na época, com procedimentos de testes de drogas menos rígidos, o medicamento foi considerado seguro após testes feitos somente em roedores, e foi prescrito inclusive à mulheres grávidas para combater enjoos matinais.
No final dos anos 1950, quando a droga já estava presente em aproximadamente 46 países, os primeiros casos de malformações congênitas foram descritos na Alemanha, Reino Unido e Austrália. Calcula-se que cerca de dez mil crianças tenham nascido em todo o mundo com deformidades físicas graves causadas pela droga, que causa uma anomalia chamada focomelia – impede que os membros superiores e inferiores se desenvolvam normalmente. Em 1962 a Talidomida foi removida da lista de remédios indicados e somente em 2010 os cientistas identificaram o mecanismo de interferência da Talidomida na formação fetal, o qual inativa a enzima cereblon, importante nos primeiros meses de vida para a formação dos membros.
A partir desse caso, a exigência de maior controle sobre a toxicidade de novas drogas em todo o mundo se estabeleceu. Uma nova droga, para ser aprovada, precisa ser testada em pelo menos três diferentes animais e também nos seres humanos, passando por pelo menos quatro fases.
Segundo Guido Lenz, professor do Departamento de Biofísica e membro do Centro de Biotecnologia da Universidade Federal do Rio Grande do Sul (UFRGS), num primeiro momento, os potenciais medicamentos devem passar por estudos pré-clínicos em modelos in vitro (enzimas, células) e in vivo (animais modelo). Após isso, devem passar por estudos clínicos fase 1 (dose e efeitos colaterais), fase 2 (primeira evidência de efeito terapêutico desejado, geralmente um número pequeno de pacientes em um único centro de pesquisa), fase 3 (multicêntrico, centenas a milhares de pacientes – procura confirmar efeito terapêutico desejado) e fase 4 (observação após comercialização).
 Foto: Crianças afetadas pela Talidomida – Leonard McCombe/Time & Life Pictures/Getty Images
Foto: Crianças afetadas pela Talidomida – Leonard McCombe/Time & Life Pictures/Getty Images
Em novembro, O Ministério da Saúde decidiu que vai testar a eficácia da substância desenvolvida na USP de São Carlos. Um grupo de estudo composto de pesquisadores ligados ao Ministério da Saúde e à Anvisa vai trabalhar em conjunto com o Instituto Nacional do Câncer e a Fiocruz para tentar comprovar a eficácia da substância. Se for aprovada, ela deverá ser produzida por laboratórios públicos oficiais. A primeira fase de testes deve ser concluída em sete meses, segundo afirmou o ministro da Ciência, Tecnologia e Inovação , Celso Pansera em coletiva de imprensa realizada após uma reunião entre o Ministério da Ciência, Tecnologia e Inovação (MCTI), o Ministério da Saúde (MS) e a Agência Nacional de Vigilância Sanitária (Anvisa). Aproximadamente R$ 10 milhões seriam destinados para as atividades ligadas à pesquisa da fosfoetanolamina em um período de 2 anos.
De acordo com o professor Lenz, o Brasil possui vários centros de pesquisa que participam de testes clínicos de em todas as fases, mas o país tem pouca experiência em coordenar testes de fase 3 e 4. “Como é uma molécula relativamente simples, os laboratórios farmacêuticos que produzem medicamentos genéricos muito provavelmente têm condições de produzir a fosfoetanolamina. Outras substâncias muitas vezes são muito complexas e requerem estruturas que não existem no Brasil para produção”.
Apesar de ser um processo caro e que demanda número grande de casos e estatísticas sofisticadas, os testes de acordo com os padrões de qualidade e controle são parte do formato “responsável” de fazer ciência mundialmente aceito, o qual permite separar efeitos — tanto positivos como negativos — de novos produtos comparados com os de placebos (substâncias sem atividade farmacológica real) ou com os de outros tratamentos mais estudados. “Da forma como está sendo feita, a distribuição é totalmente errada. Os efeitos encontrados em células e animais e os relatos de casos não são suficientes para justificar uma decisão judicial que obrigue a distribuição de uma substância sem que se passe pelos estágio de testes clínicos apropriados. Só um teste clínico bem desenhado e executado poderá dizer a eficácia e segurança deste potencial tratamento”, reitera Lenz.
O oncologista Diogo Bugano completa: “Acredito que, assim como várias outras moléculas, é algo que mostrou alguma atividade em estudos pré-clínicos de camundongos e que deve sim ser estudada, mas ainda não pode ser considerada um tratamento. Sabemos que menos de 10% das moléculas que mostram atividade em estudos pré-clínicos realmente viram tratamentos. A maioria porque – a pesar de funcionar em camundongos – não funciona em humanos; outras porque são simplesmente muito tóxicas em humanos. Não recomendo para nenhum paciente e, se forem feitas todas as etapas adequadas para o seu estudo, acredito que necessitaremos de muitos anos para poder ter a resposta definitiva se a Fosfoetanolamina funciona ou não”.
“Por outro lado, toda esta polêmica da Fosfoetanolamina está aos poucos estimulando a discussão sobre o estudo de novas drogas e o estímulo à pesquisa clínica no Brasil. Nos Estados Unidos e Europa, o uso de tratamentos experimentais – em protocolos de pesquisa – faz parte do plano terapêutico de pacientes com câncer. Mesmo em hospitais pequenos, o paciente sabe que em algum momento de seu tratamento terá a possibilidade de receber uma droga em um estudo clínico. Estes estudos são muito controlados e o paciente tem a segurança de que recebe, ao menos, o melhor tratamento padrão, além de ter a possibilidade de receber um tratamento novo que pode ser muito eficaz. Por vários motivos isso não é uma realidade no Brasil e os nossos pacientes são privados da possibilidade de participar de pesquisa clínica. Espero que essa discussão sobre a fosfoetanolamida mude a opinião pública sobre a participação em protocolos de pesquisa e nos ajude a mudar a legislação e conseguir mais verbas para pesquisa no Brasil”, conclui o oncologista.

{kind=link}